
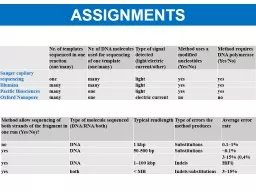
Nr of DNA molecules used for sequencing of one template onemany Type of signal detected lightelectric currentother Method uses a modified nucleotides YesNo Method requires DNA polymerase YesNo ID: 932057
Download Presentation The PPT/PDF document "ASSIGNMENTS Nr. of templates sequence..." is the property of its rightful owner. Permission is granted to download and print the materials on this web site for personal, non-commercial use only, and to display it on your personal computer provided you do not modify the materials and that you retain all copyright notices contained in the materials. By downloading content from our website, you accept the terms of this agreement.














Slide1
ASSIGNMENTS
Nr. of templates sequenced in one reaction (one/many)Nr. of DNA molecules used for sequencing of one template (one/many)Type of signal detected (light/electric current/other)Method uses a modified nucleotides (Yes/No)Method requires DNA polymerase (Yes/No)Sanger capilary sequencingonemanylightyesyesIlluminamany many lightyesyesPacific Biosciencesmany onelight yesyesOxford Nanoporemany oneelectric currentno no
Method allow sequencing of both strands of the fragment in one run (Yes/No)?
Type of molecule sequenced (DNA/RNA/both)
Typical readlength
Type of errors the method produces
Average
error
rate
no
DNA
1 kbp
Substitutions
0.1–1%
yes
DNA
50-500 bp
Substitutions
~0.1%
yes
DNA
1–100 kbp
Indels
3-15% (0,4
% HiFi
)
yes
both
<
MB
Indels
/
substitutions
3–15%
Slide2ASSIGNMENTS
Homonomy: the development of homonomous structures is governed by homologous genes originating from the common ancestor – e.g. Hox genesGene paralogy: homologues originating by gene duplication.
Slide3WHOLE GENOME COMPARISONS
DNA-DNA HYBRIDIZATIONUsed in bacterial systematicsBacterial species boundary (5% sequence difference, 30% decrease of Tm) LaboriousNxN difficultyToday substituted by Overall genome relatedness indices
Slide4ORGI (overall
genome relatedness indices) Attempts to find a simple measure of similarity by comparing whole genomes. Currently, more than 30,000 bacterial genomes. ANI (average nucleotide identity) – average identity of all homologous regions recognised by BLASTN (Goris a kol. 2007) – boundary of bacterial species 95-96%. Frequencies of 4 nukleotide „words“ (Richter a Rosseló-Móra 2009) GBDP – average genetic distance calculated from the overall alignment of two genomes (Meier-Kolthoff a kol. 2013) – bacterial species boundary 0,26. By overall alignment we mean set of alignable regions, e. g. „high-scoring segment pairs“, in BLAST. MUMi – ratio of regions with perfect match and the length of the compared genomes (Deloger a kol. 2009).
Slide5Digital
ddh
Slide6ORGI
(overall genome relatedness indices)
Slide7OTHER METHODS TO OBTAIN MOLECULAR DATA
If we had the complete genomic sequences of the organism that we want to study, we would not need any other methods. However, we do not and their sequencing is still impractically expensive for a larger number of samples. The following methods are able to quickly provide sufficient data for the questions we address in molecular phylogenetics and taxonomy.
Slide8Restri
CTION analYSESDNA isolationCutting the DNA using restriction endonucleasesThe pattern (fingerprint) is often very complexUsing low complex DNA samplesHigh quality separationDetection of only selected fragments
EcoRI
Slide9RestriCTION analYSES
Complete bacterial DNA after restriction separated on PAGE and stained by EtBrRFLP of bacterial DNAThe complexity of the bacterial genome is low, and therefore a less complex pattern is formed, from which it is possible to read individual bands
Slide10RestriCTION analYSES
VNTR – Variable Number of Tantem RepeatsUses the polymorphism in the copy number of tandem repeats – minisatelites (10-60 nt). This polymorphism is very high even in individulas of the same species.We digest the total DNA with a restriction endonuclease that does not cleave within the minisatellite, blot the digested DNA onto the membrane and hybridize to
the
labeled
probe
against
the
minisatellite
if
we
want
to
visualize
all
loci (
picture
on
the
right
),
or
against
the
minisatellite
and a
unique
sequence
in
the
neighborhood
if
we
want
to
visualize
only
one
locus
(
picture
below
).
Slide11RestriCTION analYSES
AdvantagesRelative simplicity and cheapnessDecent reproducibilityMultilocus characterSelective neutrality of charactersFor some variants of character codominance
Slide12RestriCTION analYSES
DisadvantagesThe need for more DNA The need for better and cleaner DNABetter for closely related speciesSometimes difficult to read electrophoretograms - the need to reduce the complexity of DNACharacters do not have to be independentDistance method
Slide13MiCrosatelites
Microsatelites - (also called STR - Short Tandem Repeat) are short sequence motifs (dinucleotides to hexanucleotides) occurring in some places of the genome in many tandemly arranged copies with a total length of up to 150 repeat units. On
average
,
once
every
1000
generations
,
mutations
occur
in
which
the
microsatellite
lengthens
or
shortens
,
usually
by
one
repeat
unit.
Therefore
,
there
is
a very
significant
intrapopulation
polymorphism
in
allele
length
, and
this
polymorphism
can
be
easily
studied
by PCR
amplification
of
a
given
locus
.
.
Slide14MiCrosatelites
Sliding of polymerase on the template
Slide15MiCrosatelites
Find a suitable locus (in genomic data or genomic library)Design PCR primers delimiting the locusAmplify locus Elecrophoresis of amplified fragmets (fragmentation analysis on capilary sequencher)How to proceed
Slide16MiCrosatelites
Slide17MiCrosatelites
Mendelian inheritance of microsatelites
Slide18MiCrosatelites
Intraspecific studies and studies of related speciesMendelian inheriatnece (codominance) – population geneticsPotential to analyses large amount of samplesOnly the optimalisation of the method is demanting, then easy and cheapUsage
Slide19RAPDR
andom Amplified Polymorphic DNA Amplification using short random primers
Slide20RAPD
PriceSpeedUniversalitySmall amount of DNAAdvantages
Slide21RAPD
DisadvantagesIt does not produce credible traits (many false homologies)Necessary to convert to genetic distances during analysisApplicable only to relatively related speciesMissing codominanceCharacter interferenceInfluence of DNA contaminatingLow PCR reproducibilityVariants of RAPD using primers for repetitive DNA (IRAP, REMAP, R-RAP) lower
some
disadvantages
.
Slide22AFLP
Amplified Fragment Length Polymorphism
Slide23AFLP
capilary
elektrophoresis
standard
of
molecular
weight
Amplified
fragments
Slide24AFLP
High reproducibilityHigh number of charactersMinimum of homoplasiesPossible to use character methodsAdvantagesDisadvantagesExpensive kits and instrumentationComplicated methodFor closely related species
Slide25Protein mass fingerprint
Sauer a Kliem 2010, Nature reviews microbiologyNomura 2015, BBA
Slide26Protein mass fingerprint
Slide27COMPULSORY ASSIGNMENT
+/- 0.5 point!Read this article and answer several questions.
Slide28SINE
Short INterspersed repetitive ElementsRetroposons derived from tRNA or 7 SL RNA (Alu). Size: 70-500 PB. They make up a significant portion of the eukaryotic genome, often up to 104 copies per genome. The advantage for molecular phylogenetics is that there is little chance that in two unrelated organisms they will be inserted in the same locus, and it is almost impossible to be lost from the locus without leaving a trace.
Slide29CHARACTERS AND CH. STATES
x1, y1, z1 = plesiomorphyx2 = synapomorphy for BCDy2 = autapomorphy for Bz2 = homoplazy (convergence) for ED
Slide30SINE
Find a new SINE element: searching in genomic data, random genome sequencing, screening a genomic library with a probe against SINE.Identify SINE loci that are polymorphic in the studied taxon.PCR amplification of selected locus.Verification of the presence or absence of SINE by sequencingHow to proceed
Slide31SINE
PCR productsHybridisation with SINE probeHybridisation with unique flanking sequence
Slide32SINE
Nikaido a kol. 1999
Slide33SINE
Chen a kol. 2011
Slide34BRAIN EXERCISE
The presence of a SINE element does not correspond with the known phylogeny. It is absent in chimpanzee and we know this is not the secondary loss. No experimental error involved.Try to explain it.
Slide35SINE
Applicable to higher taxa (maximum 75-100 million yearsFew characters - it is not possible to estimate the length of branchesUnique events - synapomorphy, and therefore the data can be very easily interpretedIt allows to polarize phylogeny - we know that the original state is the absence of SINEAdvantages and disadvantages
Slide36SNPs
Single Nucleotide PolymorphismA DNA polymorphism in which individuals or species differ in a single nucleotide interchangeAAGCCTAAAGCTTA In this case we speak about alleles C and T. Almost all SNPs have only 2 alleles. The genomes of two people differ by about 3 million bases (not everything is SNP).
Slide37SNPs
Slide38SNPs
Slide39SNPs
genotypizaceMolecular beacon Hybridization methods Enzymatic methods Methods based on physical features of DNAHybridization methods
Slide40SNPs –
hybridization methods906 600 lidských SNPs
Slide41SNP
s – enzymatic methodshttps://www.youtube.com/watch?v=lVG04dAAyvY&t=148s
Slide42COMPARISON OF METHODS
MetodsDNA hybridizationMass fingerptintingMicrosatelitesRFLF (VNTR)SINERAPDAFLPSNPNr. lociAllManyOneManyOneManyManyOne / manyReproducibilityVariableHighHighHighHighVariableVariableHighNature of charactersDistanceCodominanceCodominanceCodominanceRare eventDominanceCodominance?CodominanceResolutionMediumHighHighHighHighMediumHighHighUsageDifficultEasyMediumEasyMediumEasyMedium-SpeedLowHighMediumHighMediumhighHigh-