PPT-Degradazione delle macro
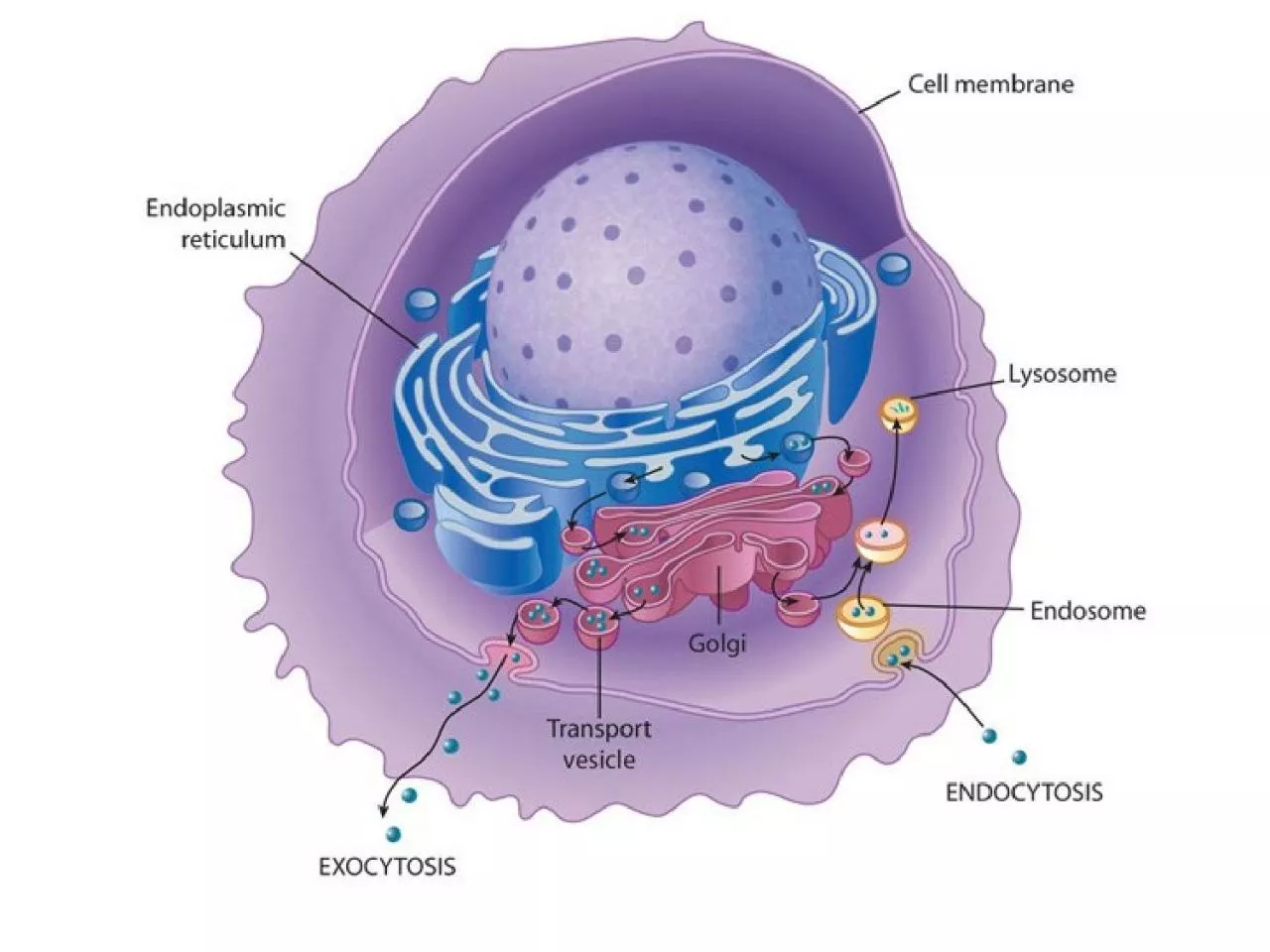
molecole EXTRACELLULARI Funzioni dei lisosomi Proteasoma degrada le proteine citosoliche Funzioni dei lisosomi Proteasoma degrada le proteine citosoliche INTRACELLULARI
Download Presentation
"Degradazione delle macro" is the property of its rightful owner. Permission is granted to download and print materials on this website for personal, non-commercial use only, provided you retain all copyright notices. By downloading content from our website, you accept the terms of this agreement.
Presentation Transcript
Transcript not available.