PPT-Decarboxylation Reaction
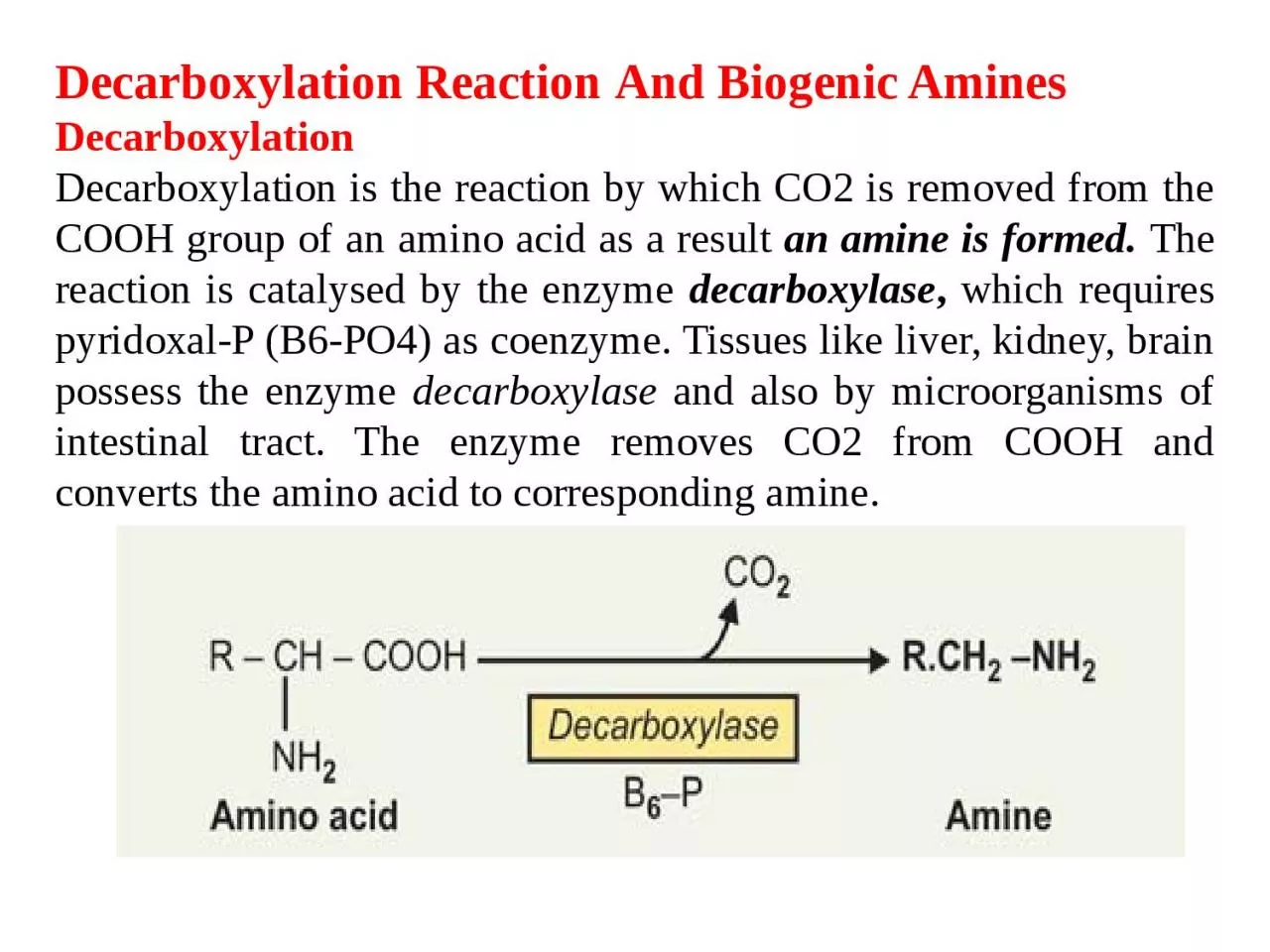
And Biogenic Amines Decarboxylation Decarboxylation is the reaction by which CO2 is removed from the COOH group of an amino acid as a result an amine is formed The
Download Presentation
"Decarboxylation Reaction" is the property of its rightful owner. Permission is granted to download and print materials on this website for personal, non-commercial use only, provided you retain all copyright notices. By downloading content from our website, you accept the terms of this agreement.
Presentation Transcript
Transcript not available.