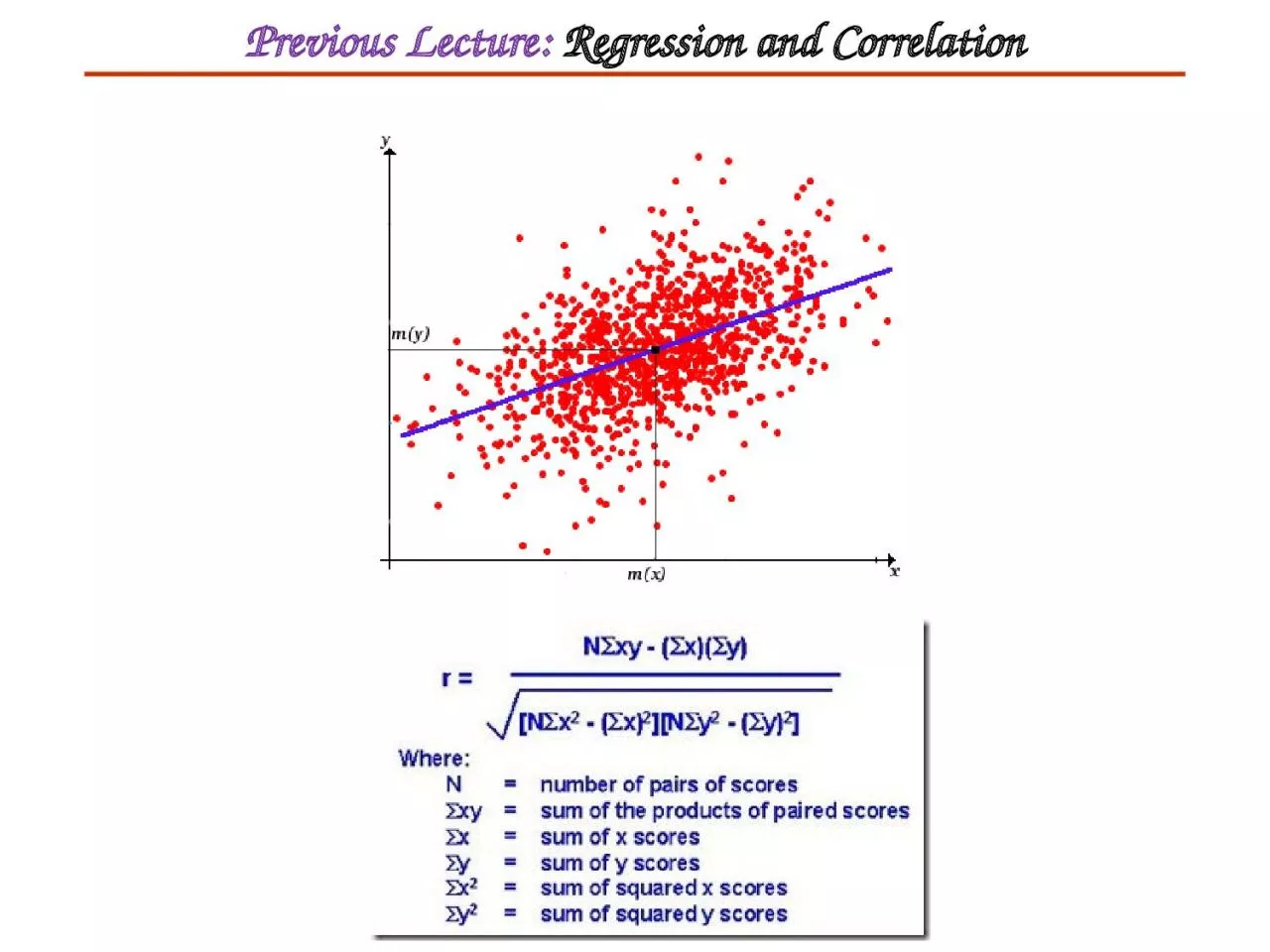
PPT-Previous Lecture: Regression and Correlation
Introduction to Biostatistics and Bioinformatics Proteomics Informatics This Lecture Proteomics Informatics Learning Objectives Structure of m ass spectrometry data
Download Presentation
"Previous Lecture: Regression and Correlation" is the property of its rightful owner. Permission is granted to download and print materials on this website for personal, non-commercial use only, provided you retain all copyright notices. By downloading content from our website, you accept the terms of this agreement.
Presentation Transcript
Transcript not available.