PPT-Lecture 4 – Characters: Molecular
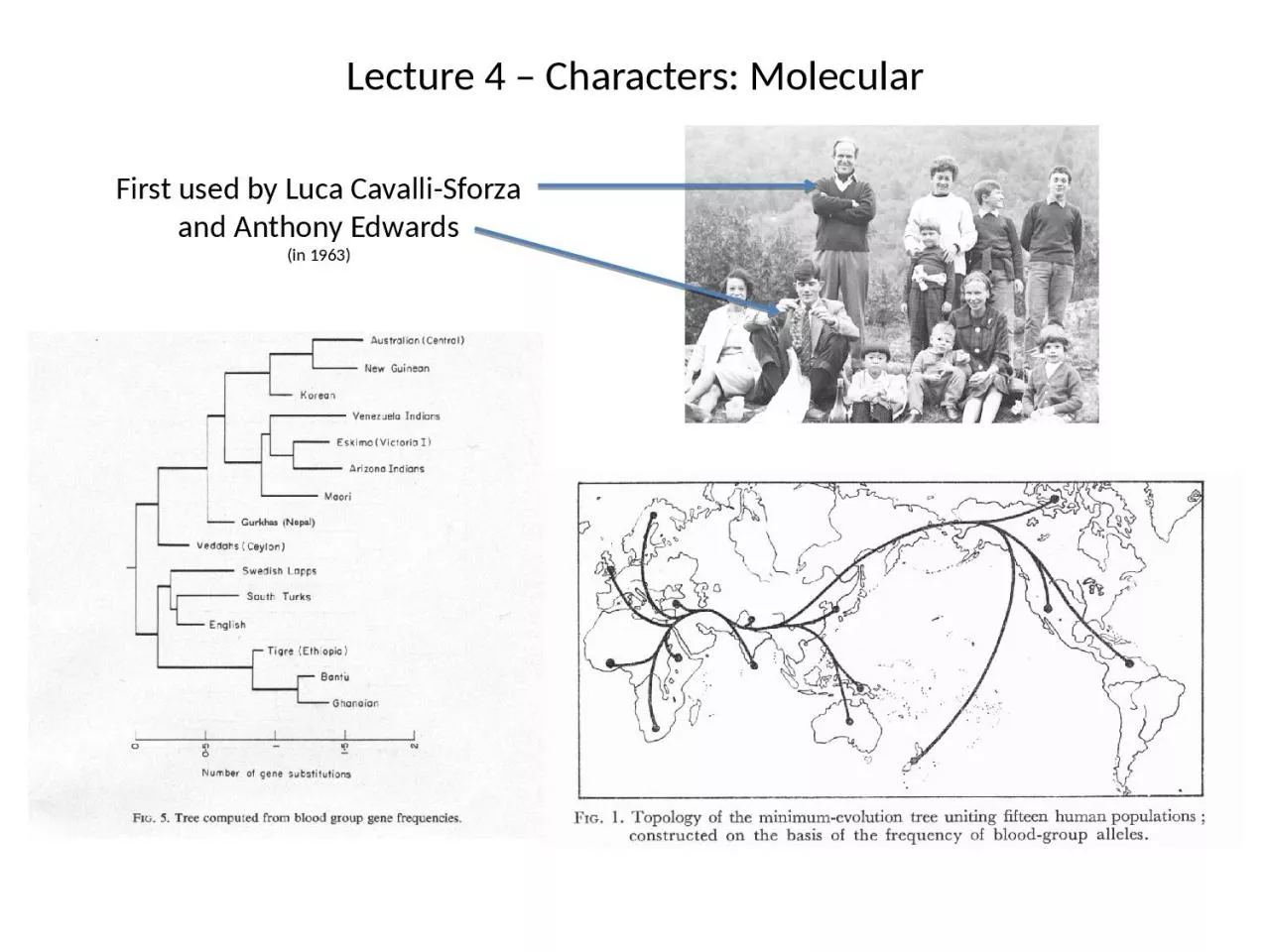
First used by Luca Cavalli Sforza and Anthony Edwards Lecture 4 Characters Molecular cwk1056 eaa292 cwk1025 eaa448 dsr5032 eaa028 fac1117 cwk1007 cwk1056
Download Presentation
"Lecture 4 – Characters: Molecular" is the property of its rightful owner. Permission is granted to download and print materials on this website for personal, non-commercial use only, provided you retain all copyright notices. By downloading content from our website, you accept the terms of this agreement.
Presentation Transcript
Transcript not available.