PPT-Fission yeast Schizosaccharomyces
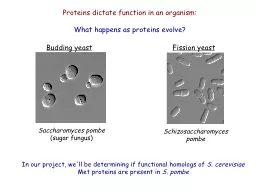
pombe Budding yeast Saccharomyces pombe sugar fungus Proteins dictate function in an organism What happens as proteins evolve In our project well be determining
Download Presentation
"Fission yeast Schizosaccharomyces" is the property of its rightful owner. Permission is granted to download and print materials on this website for personal, non-commercial use only, provided you retain all copyright notices. By downloading content from our website, you accept the terms of this agreement. Download
Presentation Transcript
Transcript not available.