PPT-MYOCARDIAL DISEASE Myocardial disease
Author : layla | Published Date : 2023-05-21
is a disaese of the myocardium that is not due to an ischaemic valvular or hypertensive heart disease It may be caused by an acute or chronic inflammatory
Presentation Embed Code
Download Presentation
Download Presentation The PPT/PDF document "MYOCARDIAL DISEASE Myocardial disease" is the property of its rightful owner. Permission is granted to download and print the materials on this website for personal, non-commercial use only, and to display it on your personal computer provided you do not modify the materials and that you retain all copyright notices contained in the materials. By downloading content from our website, you accept the terms of this agreement.
MYOCARDIAL DISEASE Myocardial disease: Transcript
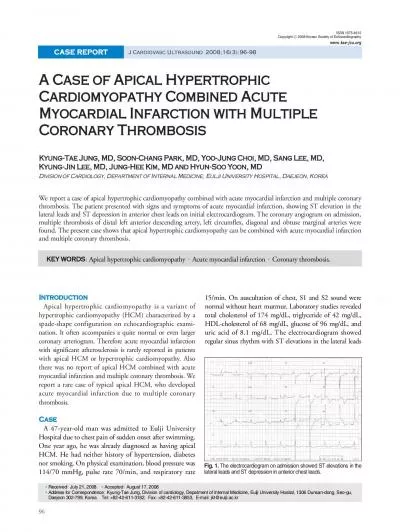
is a disaese of the myocardium that is not due to an ischaemic valvular or hypertensive heart disease It may be caused by an acute or chronic inflammatory pathology myocarditis. Susana G. Garcia MD. No Disclosure. Objectives. Review the current definition, risk factors, clinical impact and incidence of PMI. Describe the different clinical presentation of PMI and how this dictate the goal and approach to diagnosis and treatment of PMI. Byron Rudman 14019991. Brandon Wright 14015570. Brenton . Lotz. 14008442. Emilio . Guerro. 13140346. Kyle . Rorke. 13415591. Myocardial Infarction. Definition: Myocardial Infarction occurs when the heart is subject to prolonged exposure to ischemia, resulting in irreversible necrosis of the myocardium.. Coronary Heart Disease. Affects 16 million people. Causes more than 600,000 death annually. Caused by impaired blood flow to the myocardium. Accumulation of atherosclerotic plaque in the coronary arteries usual cause.. . Presented by- Dr. . Kamal. . Prakash. Sharma. . Moderator- Dr. . Manoj. Kumar . Panwar. HYPERTENSION. Hypertension is most frequent preoperative abnormality in surgical patients, with an overall prevalence of 20–25%. . B . hussam. PhD Clinical pharmacy. Coronary heart disease (CHD), sometimes described . as coronary . artery disease (CAD) or . ischaemic. heart . disease (IHD. ), is a condition in which the vascular supply to . . M.Pharm. ANGINA PECTORIS. Angina pectoris is a clinical syndrome of chest discomfort caused by reversible myocardial ischemia that produces disturbances in myocardial function without causing myocardial necrosis. . hypoperfusion. . The presence of glucose activity by FDG imaging provides evidence of viability beyond perfusion by either PET or SPECT.” . 7. PET for the Evaluation of Myocardial . Viability. Add presenter info here. infarction (STEMI). The basics. Epidemiology. Pathophysiology. Symptoms & diagnosis. Content. Epidemiology. Epidemiology. Pathophysiology. Symptoms & diagnosis. 4. CVD is a major cause of death worldwide. ArteriosclerosisTumors of blood vesselsIschemic heart diseaseValvular heart diseasesRheumatic heart diseaseInfective endocarditisMyocarditis = hardening of the arteriesAtherosclerosis Large & medium s 1 Cardiac troponins (cTn1, cTnT, high sensitivity troponins)Liver Function Tests (LFTs)Thyroid Function Tests (TFTs) Brain Natriuretic Peptides (BNP or N-terminal Electrocardiography (ECG)Chest X-R Journal of Cardiovascular Ultrasound 16 neous coronary intervention procedures, which may partially explain the lower than expected event rate in acute myocardial infarction.There was no report of api PROF. & H.O.D.. PM. Myocardial Infarction if the rapid development of myocardial necrosis by a critical imbalance between oxygen supply and demand to the myocardium. Background. Acute coronary syndromes include. ,. Associate Professor, Pathology. Sri Venkateswara Institute of Medical Sciences. Trupathi. ISCHEMIC HEART DISEASE (IHD). IHD represents a group of . pathophysiologically. related syndromes resulting from myocardial ischemia – an . Dr.Aryakrishna. A(JR1 MEDICINE). TDMC . ALAPPUZHA. The . most common reasons for which patients present . for . medical attention at ED or an OP clinic. The evaluation of . non traumatic . chest discomfort is inherently challenging owing to the broad variety of possible .
Download Document
Here is the link to download the presentation.
"MYOCARDIAL DISEASE Myocardial disease"The content belongs to its owner. You may download and print it for personal use, without modification, and keep all copyright notices. By downloading, you agree to these terms.
Related Documents