PPT-GENETIC DISORDERS DISEASES
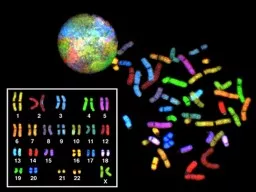
GENETIC ENVIRONMENTAL BOTH CONGENITAL HEREDITARY FAMILAL MUTATIONS PERMANENT change in DNA GENE MUTATION may and often result in a single base error CHROMOSOME MUTATION
Download Presentation
"GENETIC DISORDERS DISEASES" is the property of its rightful owner. Permission is granted to download and print materials on this website for personal, non-commercial use only, provided you retain all copyright notices. By downloading content from our website, you accept the terms of this agreement.
Presentation Transcript
Transcript not available.