PDF-A Seven novel TaySachs mutations detected by chemical mismatch cleav
Author : joanne | Published Date : 2022-10-14
starts and is usually fatal by age 4 or 5 yearsOur patient presented with early infantileonset of intractable seizures and progressive neurological deterioration
Presentation Embed Code
Download Presentation
Download Presentation The PPT/PDF document "A Seven novel TaySachs mutations detecte..." is the property of its rightful owner. Permission is granted to download and print the materials on this website for personal, non-commercial use only, and to display it on your personal computer provided you do not modify the materials and that you retain all copyright notices contained in the materials. By downloading content from our website, you accept the terms of this agreement.
A Seven novel TaySachs mutations detected by chemical mismatch cleav: Transcript
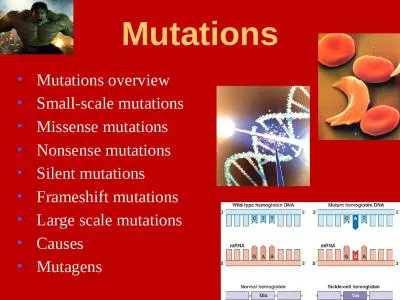
starts and is usually fatal by age 4 or 5 yearsOur patient presented with early infantileonset of intractable seizures and progressive neurological deterioration leading to death at the age of 4 yea. with dual vocational training. Karen Roiy, Senior Advisor. Agenda. The problem of skills mismatch . How to combat youth unemployment. Mobile, dynamic and open labour markets. Educational. . systems . Dr Alan Donaldson. Consultant . in Clinical Genetics. Bristol. Why do tumour analysis?. To identify 1-5% of individuals whose colon cancer may be due to Lynch syndrome, for DNA analysis.. ~15% of colon cancers are MSI high.. Guillermo . Montt. Directorate for Employment, Labour and Social Affairs. g. uillermo.montt@oecd.org. ELS Seminar. June 2015. What is field-of-study mismatch?. “English Lit – How about you?”. E.g. . Diagnosis for . Selective . Query Expansion. Le Zhao and Jamie . Callan. Language Technologies Institute. School of Computer Science. Carnegie Mellon University. Pittsburgh, PA. @SIGIR 2012, Portland, OR. Amihood Amir. Benny Porat. Moskva River. Confluence of 4 Streams. Palindrome Recognition. Approximate Matching. Interchange Matching. Online Algorithms. CPM 2014. Palindrome Recognition. - Voz'mi-ka slovo . Ian . Shuttleworth. . and Brian Foley, Queen’s University Belfast. British Society for Population Studies annual conference. 9. th. September 2015, University of Leeds. Presentation format. Context. Gender attraction errors in child . English. INTRODUCTION. Lucia . Pozzan. 1,2. , . Dorota. . Ramlogan. 3. , . and Virginia . Valian. 1,3. 1. CUNY Graduate . Center, . 2. University of Pennsylvania, . Ian Shuttleworth, David Martin and Paul Barr. S. tructure. Introduction. The data and the project. The analysis. Geography. Individual factors. Property/household factors. Concluding comments, questions and ways forward. http://io9.gizmodo.com/10-unusual-genetic-mutations-in-humans-470843733. Unharmful. Mutations. Mila . Kunis. has complete . heterochromia. . Kate Bosworth has . sectoral. . heterochromia. . Angelina Jolie has central . The sequence of bases in DNA are like the letters of a coded message or even the letters of a simple alphabet. . If we change the sequence of the letters do we change the nature of the message?. That . Small-scale mutations. Missense mutations. Nonsense mutations. Silent mutations. Frameshift mutations. Large scale mutations. Causes. Mutagens. The results of mutation. Mutations. Genetic mutations are changes in the DNA sequence, caused by various mechanisms.. Cécile Jovelet. Postdocoral. . fellow. Translational. . research. . lab. Gustave Roussy Institute, France. Introduction . 12/04/2017. 2. Lung cancer: . third most frequent cancer. Non-Small Cell Lung Cancer (NSCLC): 80% cases of lung cancer. Version 1. Zahid Atcha.. 1. Version 1. 2. What’s in this User Guide?. Version Control. Best Practice: KBD (Knowledge Based Diagnostics). Overview – ONT Mismatch. Logging In. Accessing the Journey. There are two basic types of mutations:. Gene mutations - mutations that produce changes in a single gene. Chromosomal mutations - mutations that produce changes in whole chromosomes. Gene mutations .
Download Document
Here is the link to download the presentation.
"A Seven novel TaySachs mutations detected by chemical mismatch cleav"The content belongs to its owner. You may download and print it for personal use, without modification, and keep all copyright notices. By downloading, you agree to these terms.
Related Documents